amep.spatialcor.spatialcor#
- amep.spatialcor.spatialcor(coords: ndarray, box_boundary: ndarray, values: ndarray, other_coords: ndarray | None = None, other_values: ndarray | None = None, dbin_edges: ndarray | None = None, ndists: int | None = None, chunksize: int | None = None, njobs: int = 1, rmax: float | None = None, verbose: bool = False, pbc: bool = True) tuple[ndarray, ndarray]#
Calculates the spatial correlation function of the values for atoms at positions given by coords. Distances are calculated by the cKDTree algorithm.
Notes
Works only for 2D systems in this version.
The spatial correlation function is defined by the following equation in which f can be a complex-valued scalar or vector:
- ..math::
C(r) = <f(r)*f(0)> / <f(0)^2>.
- Parameters:
coords (np.ndarray of shape (N,3)) – Coordinate frame of all particles.
box_boundary (np.ndarray of shape (3,2)) – Boundary of the simulation box in the form of np.array([[xmin, xmax], [ymin, ymax], [zmin, zmax]]).
values (np.ndarray of shape (N,) or (N,d)) – Contains per atom values for which the spatial correlation function should be calculated. For scalar values, provide a np.ndarray of shape (N,), where N is the number of particles. For vector quantities, provide a np.ndarray of shape (N,d), where d is the dimension of the vector. For example, to calculate the spatial velocity correlation function, an array of shape (N,3) would be required for a 3d system containing the speeds in each spatial dimension.
other_coords (np.ndarray of shape (M,3) or None, optional) – Coordinates to which the correlation is calculated. If None, coords is used. The default is None.
other_values (None or np.ndarray of shape (M,) or (M,d), optional) – Values corresponding to other_coords. If None, values is used. The default is None.
dbin_edges (np.ndarray, optional) – Edges of the distance bins. The default is None.
ndists (int, optional) – Number of distances. The default is None.
chunksize (int, optional) – Divide the system into chunks of this size to save memory during the processing (the value gives the number of atoms for each chunk; it is suggested to use values between 1000-5000). The default is None.
njobs (int, optional) – Number of jobs used for parallel computation. The default is 1.
rmax (float, optional) – Maximal distance to consider. The default is None.
verbose (bool, optional) – If True, additional information is printed and a progress bar is shown. The default is False.
pbc (bool, optional) – If True, periodic boundary conditions are applied. The default is True.
- Returns:
np.ndarray of shape (ndists,), dtype float – Averaged correlation function.
np.ndarray of shape (ndists,), dytpe float – Distances.
Examples
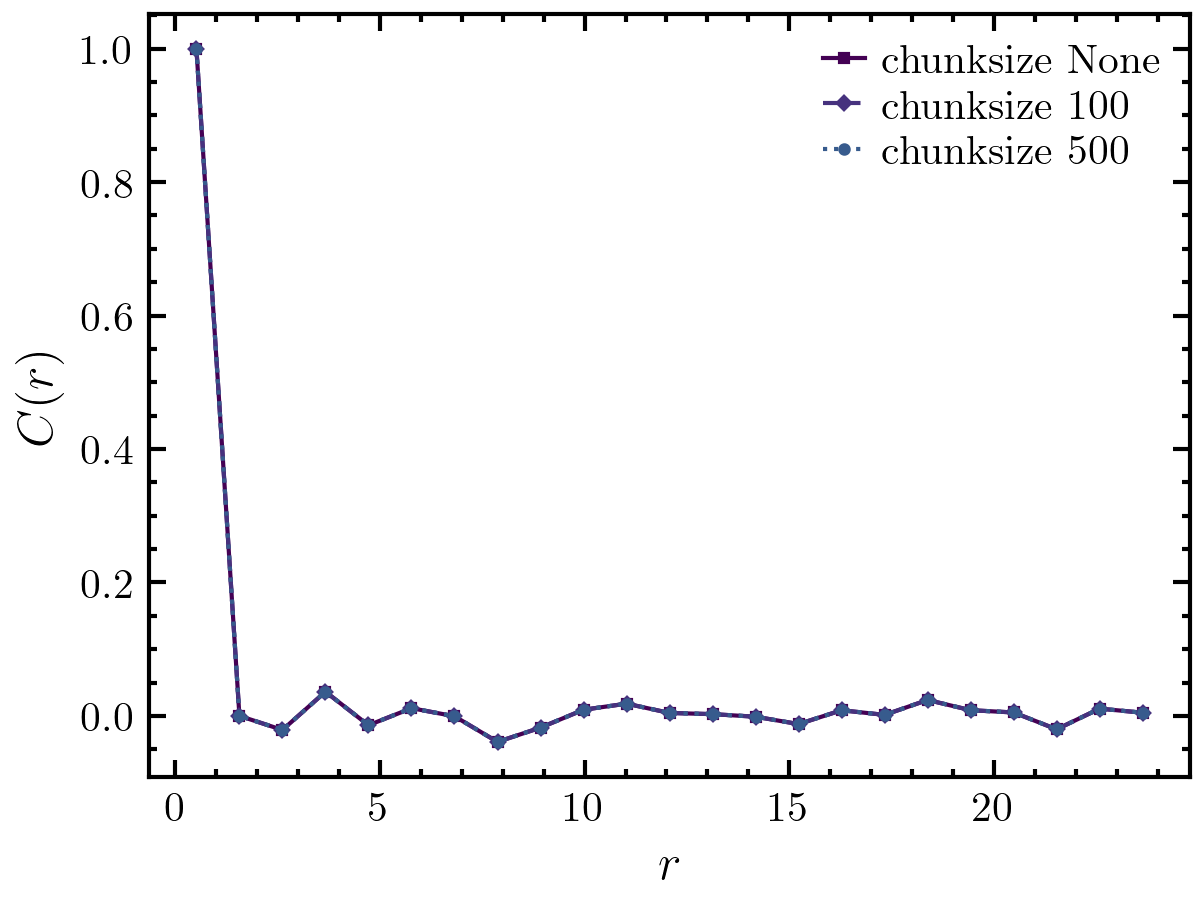
>>> import amep >>> traj = amep.load.traj("../examples/data/lammps.h5amep") >>> frame = traj[-1] >>> cor0, d0 = amep.spatialcor.spatialcor( ... frame.coords(), frame.box, frame.data('vx'), ... chunksize=None, njobs=4, rmax=25, verbose=True ... ) >>> cor1, d1 = amep.spatialcor.spatialcor( ... frame.coords(), frame.box, frame.data('vx'), ... chunksize=100, njobs=4, rmax=25, verbose=True ... ) >>> cor2, d2 = amep.spatialcor.spatialcor( ... frame.coords(), frame.box, frame.data('vx'), ... chunksize=500, njobs=4, rmax=25, verbose=True ... ) >>> fig, axs = amep.plot.new() >>> axs.plot(d0, cor0, label='chunksize None', ls='-') >>> axs.plot(d1, cor1, label='chunksize 100', ls='--') >>> axs.plot(d2, cor2, label='chunksize 500', ls=':') >>> axs.set_xlabel(r'$r$') >>> axs.set_ylabel(r'$C(r)$') >>> axs.legend() >>> fig.savefig('./figures/spatialcor/spatialcor-spatialcor.png') >>>